
Int J Chem Res, Vol 5, Issue 2, 1-4Research Article
SYNTHESIS AND CHARACTERIZATION OF BOC-PROTECTED THIO-1,3,4-OXADIAZOL-2-YL DERIVATIVES
S. SYED SHAFI1*, R. RAJESH1,2, S. SENTHILKUMAR2
1Department of Chemistry, Thiruvalluvar University, Serkkadu, Vellore, India, 2Department of Chemistry, Arunai Engineering College, Tiruvannamalai, India
*Email: [email protected]
Received: 18 Mar 2021 Revised and Accepted: 10 Jun 2021
ABSTRACT
Objective: The objective of this work is to synthesis thio-1,3,4-oxadiazol-2-yl derivatives for industrial applications.
Methods: The synthesis was done by the reaction of (S)-2-amino-2-phenylacetic acid with ethanol-derived ethyl (S)-2-amino-2-phenylacetate. The reaction of ethyl (S)-2-amino-2-phenylacetate with Boc anhydride and hydrazine produces ethyl (S)-2-((tert-butoxycarbonyl)amino)-2-phenylacetate. The intermediate 5-alkyl amino-1,3,4-oxadiazole-2-thiols have been isolated as stable compounds.
Results: To study the characteristics of these compounds, they are subjected to 1HNMR, 13CNMR, and IR. These studies examine the chemical structure of synthesized compounds. The mass of the novel compounds was established with the help of the LCMS test. The structural and optical properties were examined by the powder XRD and photoluminescence spectrum respectively.
Conclusion: The sharp intense peak in the powder XRD indicates the crystal nature of the sample. The values obtained from the LCMS test indicate that the compounds have good thermal stability. The absorption peak at 358 nm in the PL spectrum indicates that the sample can be used in photoelectronic devices.
Keywords: (S)-2-amino-2-phenylacetic acid, Crystallization, Optical activity
© 2021 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/)
DOI: http://dx.doi.org/10.22159/ijcr.2021v5i3.169. Journal homepage: https://ijcr.info/index.php/journal
INTRODUCTION
The heterocyclic compounds have biological and pharmaceutical applications. So, they attract researchers to synthesize heterocyclic compounds for diverse applications. These heterocyclic compounds can also be synthesized from amino acids. The peptides are the important constituents of living organisms possessing diverse biological functions. But its applications were limited because of its poor metabolic stability in vivo, low oral bioavailability, and hydrolysis by proteases. These drawbacks can be overcome by its reaction with various amide-surrogates such as urea, thiourea, carbamate, and heterocycles and thus making them suitable for various applications [1, 2]. In particular, the insertion of heterocycles such as tetrazole, triazole, thiazole, and isoxazoline in the place of amide bond is of considerable interest in designing the peptidomimetics due to the added pharmacophoric values of those aromatic nuclei [3, 4]. The compounds possessing 1,3,4-oxadiazoles belong to a group of heterocycles. These heterocycles have potential applications in pharmaceutical applications and gained a special interest in drug discovery [5]. Many of them exhibit antibacterial, anticonvulsant, anticancer activities and are used to fight infections involving AIDS [6-8]. This heterocycle can also be used as a surrogate of amide and esters [9]. In agriculture, these compounds are used as herbicides, fungicides, and insecticides [10]. The1,3,4-oxadiazoles possess photochemical, photophysical, electrochemical properties and also have thermal and electroluminescent properties [11-13]. The 1,3,4-oxadiazoles are used to produce organic light-emitting diodes (OLED), optical brighteners, and laser diodes in electronics [16, 17]. The 1,3,4-oxadiazoles derivatives exhibit the crystal structure [16-18]. Based on earlier studies, an attempt is made to prepare thio-1,2,3-oxadiazole derivatives. The prepared samples were characterized for their potential applications.
MATERIALS AND METHODS
Materials
The chemicals L-phenyl glycine, potassium carbonate, di-tert-butyl dicarbonate, hydrazine hydrate, carbon disulphide, potassium hydroxide, dimethylformamide(DMF), ethyl acetate, substituted halides were purchased from Sigma-Aldrich and used without purification. The dry ethyl acetate, hexane, and ethanol were obtained from Spectrochem for the crystallization process.
Instruments and methods
To obtain the information about the functional groups present the samples were undergone FTIR spectral studies using the Perkin-Elmer spectrum 100 series spectrophotometer. The 1HNMR spectra were recorded on a 400MHz Varian spectrometer to get the number of protons and their position. The 13CNMR spectra of these compounds were taken for the samples with the help of a 100MHz Brucker spectrometer with TMS as internal standard.
The mass spectra are recorded on Shimadzu mass spectrometer. All the reactions were monitored by TLC plates and their spots were visualized by exposing them to a UV lamp, iodine chamber, or KMnO4, and it was performed with silica gel 60-120mesh. To ensure the crystalline nature of the sample powder XRD was taken from the XPERT-PRO-Gonio scan-2 m instrument. The photoluminescence property of the samples was tested by Cary Eclipse-EL08083851 photometer. The elemental analysis was done by the Varian instrument (VARIO EL-3 series analyzer).
Synthesis and characterization
Synthesis of ethyl(S)-2-amino-2-phenylacetate (2)
About 100 ml of ethanol was taken in a round bottom flask. In this, 10 g (66 mmol) of phenyl glycine was added and stirred well. Then con. H2SO4 7 ml (0.2 eq, 13 mmol) was added gradually. This solution is maintained at a temperature not exceeding 35 °C. Then, the reaction mixture was refluxed for 16 h. The mixture was monitored by TLC (mobile phase Ethyl acetate: hexane: 8:2). The reaction mixture was concentrated, washed with water, and dried with Na2SO4. It results in an oily residue. This ester was directly used for the second stage without carrying for any further purification.
It was obtained as colourless liquid. The yield was 66 %. (LCMS: 95.2 % purity). B. pt.104-106 °C, IR (KBr, cm-1): vmax 1740 (C=O), 1537-1481(C=C), 3342 (amide-NH2), 1259-1043 (C-O, C-N), 982-726 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.321-1.338 (t, 3H, J=6.8Hz,-CH3), 4.894-4.914 (t, 1H, J=8 MHz,-CH), 3.964-4.142 (m, 2H,-CH2), 7.233-7.314 (m, 5H, Ar-CH), 8.563-8.578 (d, 2H, J=6Hz,-NH2). 13CNMR (100 MHz, DMSO-d6, ppm): δ 15.16, 64.70, 128.32, 129.78, 128.63, 136.20, 176.40, 63.82. For C10H13NO2, Calculated: C-67.02 %, H-7.31 %, N-7.82 %, O-17.85 %. Found: C-67.14 %, H-7.26 %, N-7.64 %, O-17.96 %. LCMS [M+1]+: m/z 180.6.
Synthesis of ethyl(S)-2-((tert-butoxy carbonyl)amino)-2-phenylacetate(3)
The appropriate phenylglycine ethyl ester 8 g (44 mmol) and 1.4 ml (10.6 mmol) of triethylamine was stirred for about 20 min. Then 3.28 g (0.3 eq, 13 mmol) of di-tert-butyl dicarbonate was added and left agitating at room temperature for 24 h. The triethylamine was precipitated and appear as white. It was filtered off and the solution was evaporated on the rotary evaporator. The white crude product was dried in air and crystallized from isopropanol.
It was obtained as white solid with 52 % yield, (LCMS: 95.7 % purity), m. pt.112-114 °C, IR (KBr, cm-1): vmax 1682(C=O), 1526-1493 (C=C), 3346 (amide-NH), 1262-1036 (C-O C-N), 994-713 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.408 (s, 9H,-CH3), 7.257-7.326 (m, 5H, Ar-CH), 5.527-5.545 (d, 1H, J=7.2MHz,-CH), 8.324-8.340 (d, 1H, J=6.4Hz,-NH), 4.142-4.379 (m, 2H,-CH2), 1.328-1.346 (t, 3H, J=7.2Hz,-CH3). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.56, 79.58, 170.36, 62.74, 129.45, 128.98, 127.38, 136.82, 62.82, 176.32, 2.82, 16.72. For C15H21NO4, Calculated: C-64.62 %, H-7.42 %, N-5.22 %, O-22.74 %. Found: C-64.50 %, H-7.58 %, N-5.01 %, O-22.91 %. LCMS [M+1]+: m/z 280.7.
Synthesis of tert-butyl(2-hydrazineyl-2-oxo-1-phenyl)ethyl)carbamate(4)
In 20 ml of ethanol, the compound N-protected phenylglycine ethyl ester (3) 6.4 g (24 mmol) was dissolved and then 3.6 ml of 98 % hydrazine hydrate (76 mmol) was dropped in. It was stirred for 24 h and concentrated under reduced pressure. The oily residue was crystallized by trituration with 10 ml of hexane. The crude product was filtered off and recrystallized from a mixture of hexane–ethanol (2:1, v/v).
It was obtained as white solid in 67 % of yield, (LCMS: 95.3 % purity), m. pt.96-98 °C, IR (KBr, cm-1): vmax 1623(C=O), 1593-1518 (C=C), 3340 (amide-NH), 1309-1024 (C-O, C-N), 1000-675 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.380 (s, 9H,-CH3), 2.508-2.526 (d, 1H, J=7.2Hz,-CH), 5.123-5.145 (d, 1H, J=8.8MHz,-CH), 7.227-7.338 (m, 5H, Ar-CH), 4.261-4.276 (d, 3H, J=6Hz,-NH), 9.351-9.368 (t, 1H, J=6.8Hz,-NH). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.62, 78.85, 169.88, 56.77, 128.65, 127.48, 127.93, 139.41, 19.88. For C13H19N3O3 Calculated: C-58.85 %, H-7.22 %, N-15.84 %, O-18.09 %. Found: C-58.57 %, H-7.43 %, N-15.68 %, O-18.33 %. LCMS [M+1]+: m/z.266.30.
Synthesis of tert-butyl (S)-(5-mercapto-1,3,4-oxadiazol-2-yl)(phenyl)methylcarbamate (5)
To a stirred solution of tert-butyl(2-hydrazineyl-2-oxo-1-phenyl)ethyl)carbamate (4) (5 g, 18.8 mmol), in ethanol (30 ml), KOH 2.1 g (37.6 mmol) were added and stirred for 30 min. Then CS2 1.75 g (22.56 mmol) was added and stirred for 1 h at room temperature. After completion of the reaction, concentrated to the residue, acidify with 1.5N HCl, solid was thrown out, filtered, and dried.
It was obtained as white solid with 67 % of yield (LCMS: 95.3 % purity), m. pt.104-106 °C, IR (KBr, cm-1): vmax 1681(C=O), 1529-1518 (C=C), 3350 (amide-NH), 1016-1245 (C-O C-N), 991-704 (C-H), 637 (C-S). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.410 (s, 9H,-CH3), 8.198-8.216 (d, 1H, J=7.2Hz-NH), 6.156-6.175 (d, 1H, J=7.6Hz-CH), 7.298-7.434 (m, 5H, ArH), 12.908 (s, 1H,-SH). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.46, 79.23, 155.82, 59.23, 126.68, 128.26, 127.03, 141.96, 163.32. For C14H17N3O3S, Calculated: C-54.70 %, H-5.58 %, N-13.67 %, O-15.62 %, S-10.43 %. Found: C-54.64 %, H-5.46 %, N-13.75 %, O-15.67 %, S-10.48 %. LCMS [M+1]+: m/z 308.8.
Preparation of 1,3,4-oxadiazol derivatives (6a-d)
The one equivalent (100 mg) weighed compound-(5) was dissolved in DMF (2 ml), K2CO3 (2 eq) was added and the reaction mixture was stirred for 15 min. Then alkyl/aryl halide (1 eq) solution was added dropwise and stirred for 16 h at room temperature. After the completion of the reaction (monitor by TLC), ice water was added and extracted with ethyl acetate. The organic layer was filtered to remove potassium carbonate. The filtrate was concentrated and diluted with Ethyl acetate (2x 2 ml) washed with water and brine solution, dried, and concentrated. The crude was purified by column chromatography (gradient elution of 30-40 % of ethyl acetate in hexane) to get desired product (6a-d).
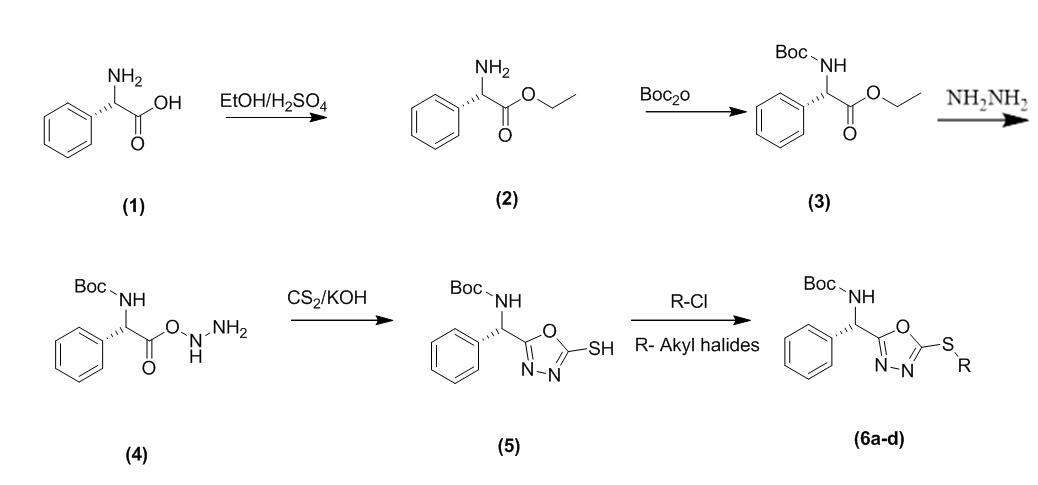
Fig. 1: The scheme of reaction
Table 1: List of alkyl halides used in the reaction
| Compound | Alyl halides | Product | Yield % | m.pt. ᵒC |
| 6a | 4-fluorobenzyl chloride | 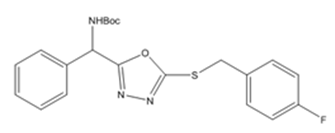 |
95 | 96-97 |
| 5b | Benzyl bromide | 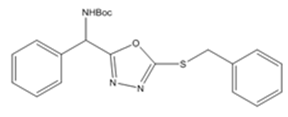 |
92 | 109-111 (b.pt) |
| 6c | Propyl iodide | 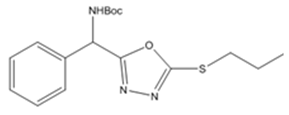 |
95 | 68-70 |
| 5d | 3-fluoro-5-(trifluoromethyl) benzyl bromide |  |
95 | 98-100 |
Synthesis of tert-butyl((5-((4fluorobenzyl)thio)-1,3,4-oxadiazol-2-yl)(phenyl)methyl) carbamate (6a)
The aryl halide 4-fluoro benzyl chloride was added in this reaction. It gives the compound 6(a) which was also a white solid with 95 % yield. (LCMS: 95.2 % purity), m. pt. 96-97 °C, IR (KBr, cm-1): vmax 1694 (C=O), 1577-1476 (C=C), 3396 (amide-NH), 1240-1007 (C-O, C-N), 626(C-S), 1007-1055 (C-F), 969-692 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.478 (s, 9H,-CH3), 8.252-8.259 (d, 1H, J=2.8Hz,-NH), 5.754-5.772 (d, 1H, J=7.2Hz,-CH), 7.080-7.121 (m, 5H, ArH), 4.451 (s, 2H,-CH2), 7.373-7.420 (m, 4H, ArH). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.5, 79.53, 155.36, 51.00, 128.77, 129.08, 127.97, 137.22, 163.24, 35.45, 131.59, 129.08, 115.67, 160.81. For C21H22FN3O3S, Calculated: C-60.92 %, H-5.47 %, F-4.76 %, N-10.04 %, O-11.29 %, S-7.52 %. Found: C-60.71 %, H-5.34 %, F-4.57 %, N-10.11 %, O-11.55 %, S-7.72 %. LCMS [M+1]+: m/z 416.7.
Synthesis of tert-butyl((5-(benzylthio)-1,3,4-oxadiazol-2-yl)(phenyl) methyl) carbamate (6b)
To get the compound 6(b), benzyl bromide (aryl halide) was added and results colour less liquid with 92 % yield. (LCMS: 95.5 % purity), b. pt.109-111 °C, IR (KBr, cm-1): vmax 1703 (C=O), 1579-1454 (C=C), 3311 (amide-NH), 1276-1016 (C-O, C-N), 605 (C-S) 977-696 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.393 (s, 9H,-CH3), 8.254-8.274 (d, 1H, J=8Hz,-NH), 6.054-6.075 (d, 1H, J=8.4Hz,-CH), 7.326-7.401 (m, 5H, ArH), 4.573 (s, 2H,-CH2), 7.543-7.707 (m, 5H, ArH). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.56, 79.50, 155.37, 51.01, 128.75, 129.04, 127.93, 137.26, 163.98, 35.01, 137.26, 128.75, 129.04, 127.93. For C21H23N3O3S, Calculated: C-63.46 %, H-5.83 %, N-10.57 %, O-12.07 %, S-8.07 %. Found: C-63.21 %, H-5.95 %, N-10.39 %, O-12.02 %, S-8.43 %. LCMS [M+1]+: m/z.398.9.
Synthesis of tert-butyl((5-(propylthio)-1,3,4-oxadiazol-2-yl)(phenyl)methyl)carbamate (6c)
The reaction of propyl iodide (alkyl halide) and the compound-5 results the compound 6(c) in the form of white solid. Its yield percentage is 95 %. (LCMS: 94.4 % purity), m. pt.68-70 °C, IR (KBr, cm-1): vmax 1685 (C=O), 1577-1475 (C=C), 3361 (amide-NH), 11286-1020 (C-O, C-N), 633(C-S) 969-702 (C-H). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.400 (s, 9H,-CH3), 8.250-8.257 (d, 1H, J=2.8Hz,-NH), 6.054-6.059 (d, 1H, J=2Hz,-CH), 7.352-7.441 (m, 5H, ArH), 3.320-3.339 (t, 2H, J=7.6Hz,-CH2), 1.650-1.688 (m, 2H,-CH2), 0.860-0.887 (t, 3H, J=10.8Hz,-CH3). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.50, 79.42, 155.30, 51.10, 128.70, 129.01, 128.03, 137.28, 164.50, 31.98, 21.24, 13.96. For C18H25N3O3S, Calculated: C-58.43 %, H-6.63 %, N-12.02 %, O-13.74 %, S-9.18 %. Found: C-58.54 %, H-6.52 %, N-12.14 %, O-13.68 %, S-9.12 %. LCMS [M+1]+: m/z 351.0.
Synthesis of tert-butyl((5-((3-fluoro5-(trifluoromethyl)benzyl)(thio)-1,3,4-oxadiazol-2-yl) (phenyl) methyl) carbamate(6d)
The aryl halide 3-fluoro-5-(trifluoro methyl) benzyl bromide was added and reacted to produce the compound 6(d). It was obtained as white crystalline solid with 95 % yield. (LCMS: 95.6 % purity), m. pt.98-100 °C, IR (KBr, cm-1): vmax 1681 (C=O), 1573-1473 (C=C), 3350 (amide-NH), 1292-1022 (C-O, C-N), 615(C-S) 974-694 (C-H), 1022-1049 (C-F), 1049-1165 (C-F, CF3). 1HNMR (400 MHz, DMSO-d6, ppm): δ 1.404 (s, 9H,-CH3), 8.284-8.304 (d, 1H, J=8Hz,-NH), 6.078-6.099 (d, 1H, J=8.4Hz,-CH), 7.353-7.430 (m, 5H, ArH), 4.460 (s, 2H,-CH2), 7.268-7.315 (m, 3H, ArH). 13CNMR (100 MHz, DMSO-d6, ppm): δ 28.57, 79.50, 155.37, 51.01, 128.20, 128.75, 127.99, 137.26, 163.98, 36.26, 137.26, 121.36, 129.43, 110.56, 163.98, 119.24, 124-48. For C22H21F4N3O3S, Calculated: C-54.65 %, H-4.38 %, F-15.72 %, N-8.69 %, O-9.93 %, S-6.63 %. Found: C-54.82 %, H-4.65 %, F-15.53 %, N-8.82 %, O-9.76 %, S-6.42 %. LCMS [M+1]+: m/z.484.4.
All the reaction schemes were shown in fig. 1. The yield and melting point of these samples are presented in table 1.
RESULTS AND DISCUSSION
Powder XRD studies
The powder XRD pattern is shown in fig. 2. From the graph, it is observed that the peaks are sharp and intense. This shows that the sample is pure and in crystalline nature.
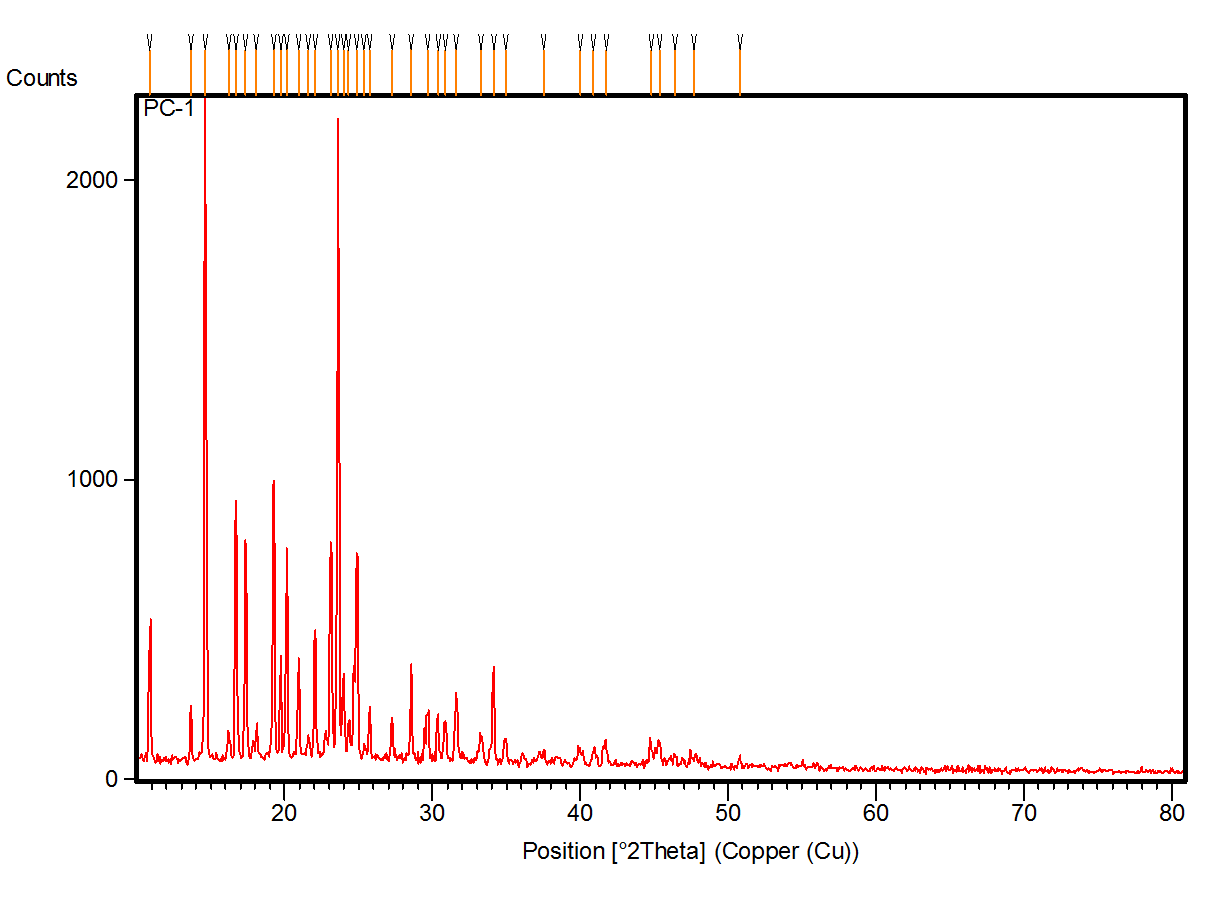
Fig. 2: Tert-butyl (2-hydrazineyl-2-oxo-1-phenyl) ethyl) carbamate (4)
The crystalline size is calculated from the Debye-scherrer formula

λ → wavelength 1.546 A°
β → Full width half (0.1476 deg = 0.002576 rad)
θ → Angle of diffraction (14.6486/2 = 7.3243 deg = 0.127833 rad)
D = 0.9 X 0.154/0.002576 X Cos (0.1278)
D = 54.2658 nm.
The powder XRD pattern is shown in fig. 3. From the graph, it is observed that the peaks are sharp and intense. This shows that the sample is pure and in crystalline nature.
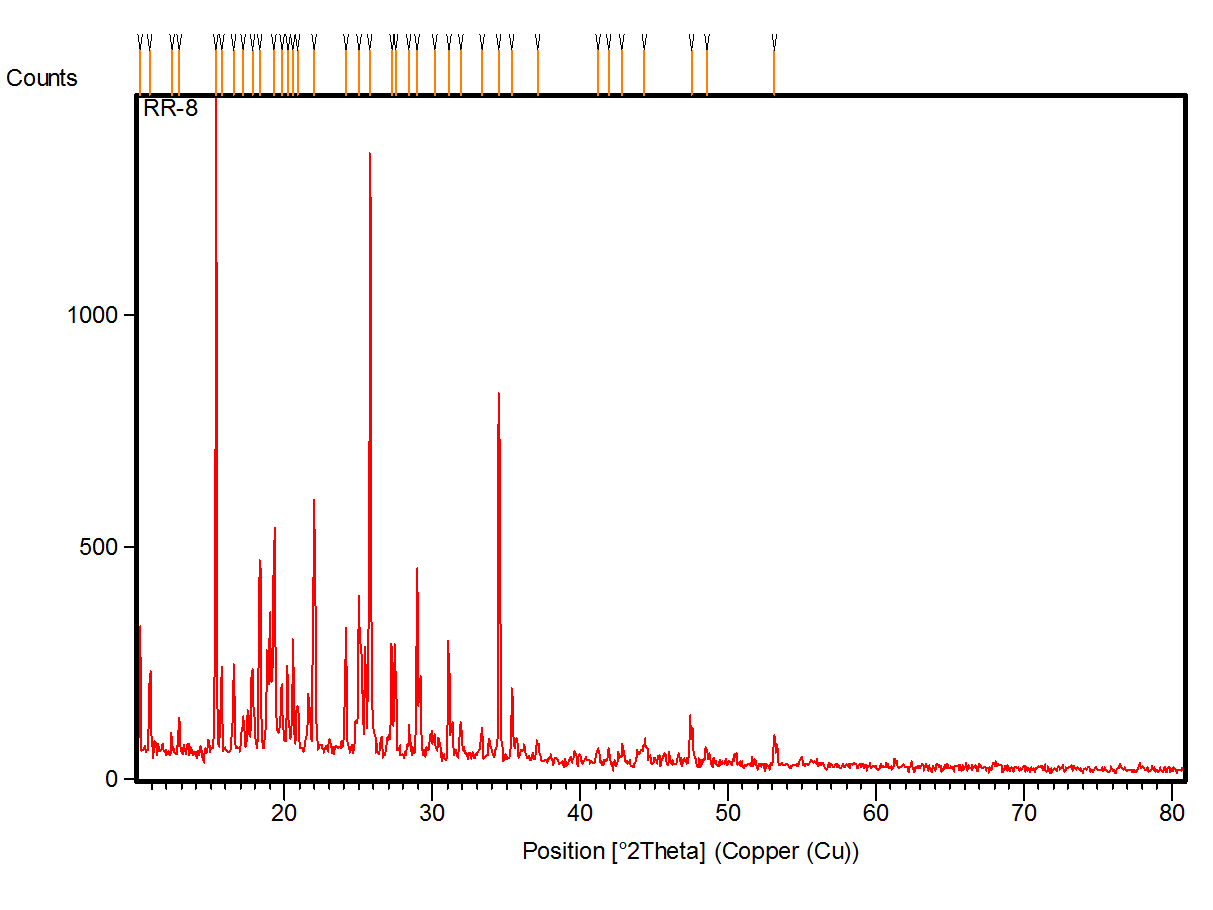
Fig. 3: Synthesis of tert-butyl((5-((3-fluoro5-(trifluoromethyl)benzyl)(thio)-1,3,4-oxadiazol-2-yl) (phenyl) methyl) carbamate (6d)
The crystalline size of the samples are calculated and presented in table 2
Table 2: Crystalline size of the compounds
| S. No. | Compound name | Size (nm) |
| 1 | 4 | 54.2658 |
| 2 | 6d | 54.3108 |
Generally, the crystalline size value will increase or decrease depending on the ordering inside the material. So when materials are added it will disturb the order of the parent compound also the additional compound produces strain inside the crystal. These strains restrict the growth of the crystal and hence result in smaller size crystals. The incorporation of additional compounds is evident from the decrease in the crystalline size as indicated in the table.
Photoluminescence
The photoluminescence (PL) spectrum examines material for its wide applications in the field of medical, biochemical, and chemical research. In PL spectroscopy, generally, a beam of light excites the electrons in the molecule of given materials and causes them to emit light in a longer wavelength than the observed radiation. The fig. Fig. 4-5 show the PL spectra of the samples. These spectra give the absorption wavelength at 358 nm which means the emission of blue radiation. The absorption peak is due to the band-to-band electronic transition in a material. When the PL spectrum of compound 6d is compared with compound 4, it is observed that the intensity of the peaks is high and also few less intense peaks were seen. These additional peaks are due to the electronic transition between the new electronic levels caused by the addition compound. The result predicts the use of the materials as a color filter.
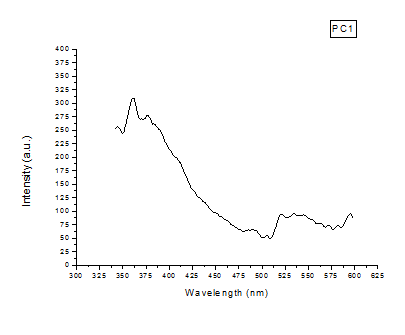
Fig. 4: PL spectrum of the compound.4
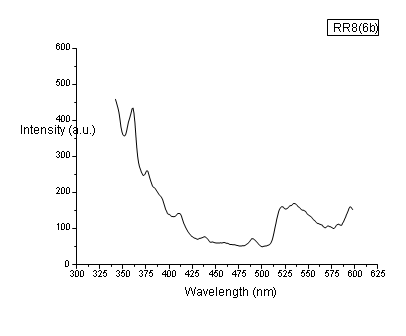
Fig. 5: PL spectrum of the compound.6d
CONCLUSION
The synthesis of derivatives of thiol-1,2,3-oxadiazole (6a-d) was carried out. The functional groups present in the samples were studied from the FTIR spectra and thus it confirms the synthesis of the compounds. The proton and carbon positions of these were obtained through 1HNMR and 13CNMR spectra respectively. The LCMS study indicates the good yield of all the compounds. The melting point of the samples was studied and shows that almost all the compounds except 6(c) have high melting points. It means that the aliphatic compounds have lower melting point compared with aromatic compounds. The crystalline nature of these samples 6 (d) was confirmed by the powder X-ray diffraction studies. These samples show less optical nature as studied from PL study.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
All the authors have contributed equally.
CONFLICT OF INTERESTS
Declared none
REFERENCES
Emmons TK, Murali R, Greene MI. Therapeutic peptides and peptidomimetics. Curr Opin Biotech 1997;8:435–41.
Gellman SH. Foldamers: a manifesto. Acc Chem Res 1998;31:173–80.
Yu KL, Johnson RL. Synthesis and chemical properties of tetrazole peptide analogs. J Org Chem 1987;52:2051–9.
Tam A, Arnold U, Soellner MB, Raines RT. Protein prosthesis: 1,5-disubstituted-[1,2,3]-triazoles as cis-peptide bond surrogates. J Am Chem Soc 2007;129:12670–71.
Rostom SAF, Shalaby MA, El-Demellawy MA. A novel class of potential antitumor and anti-HCV agents. Eur J Med Chem 2003;38:959–74.
Ogata M, Atobe H, Kushida H, Yamamoto K. In vitro sensitivity of mycoplasmas isolated from various animals and sewage to antibiotics and nitrofurans. J Antibiot 1971;24:443.
Schlecker R, Thieme PC. The synthesis of antihypertensive 3-(1,3,4-oxadiazol-2-yl)phenoxypropanolahines. Tetrahedron 1988;44:3289.
Kohara Y, Imamiya E, Kubo K, Wada T, Inada Y, Naka T. A new class of angiotensin II receptor antagonists with a novel acidic bioisostere. Bioorg Med Chem Lett 1995;5:1903.
Andersen KE, Lundt BF, Jorgensen AS, Braestrup C. Oxadiazoles as bioisosteric transformations of carboxylic functionalities. Eur J Med Chem 1996;31:417-25.
Zou XJ, Lai LH, Zhang ZX. Synthesis, fungicidal activity, and 3D-QSAR of pyridazinone-substituted 1,3,4-oxadiazoles and 1,3,4-thiadiazoles. J Agric Food Chem 2002;50:3757–60.
Wang C, Jung GY, Hua Y, Pearson C, Bryce MR, Petty MC, et al. The effects of processing conditions on the efficiency and lifetime of organic light emitting devices incorporating a new oxadiazole derivative. Chem Mater 2001;13:1167.
Andrea Pace, Ivana Pibiri, Silvestre Buscemi, Nicolo Vivona. Photochemistry of fluorinated heterocyclic compounds. an expedient route for the synthesis of fluorinated 1,3,4-oxadiazoles and 1,2,4-triazoles. J Org Chem 2004;69:4108-15.
Agnieszka Kudelko, Monika Wroblowska, Tomasz Jarosz, Katarzyna Laba, Mieczysław Lapkowski. Synthesis, spectral characteristics and electrochemistry of symmetrically substituted hybrids derived from 2,5-bis(4-bromophenyl)-1,3,4-oxadiazole under suzuki cross-coupling reaction. ARKIVOC 2015;287-302.
Zhang X, Tang B, Zhang P, Li M, Tian W. Synthesis and characterization of 1,3,4-oxadiazole derivatives containing alkoxy chains with different lengths. J Mol Struct 2007;846:55-64.
Zhi-Kuan Chen, Hong Meng, Yee Hing Lai, Wei Huang. Photoluminescent poly(p-phenylenevinylene)s with an aromatic oxadiazole moiety as the side chain. Macromolecules 1999;32:4351-8.
Li-Ping Han, Bin Lib, Jie Liu. N-Phenyl-N-{4-[5-(4-pyridyl)-1,3,4-oxadiazol-2-yl]phenyl}aniline. Acta Cryst 2008;E64:O242.
Kudelko A, Zielinski W. An efficient synthesis of new 2-aminomethyl-1, 3, 4-oxadiazoles from enantiomeric phenylglycine hydrazides. Tetrahedron 2009;65;1200-6.
Vommina V, Sureshbabu B, Vasantha G, Nagendra. A facile one-pot synthesis of Na-Z/Boc-protected S-linked 1,3,4-oxadiazole tethered peptidomimetics. Tetrahedron Lett 2012;53:1332-6.